![]() https://doi.org/10.35845/kmuj.2024.23635 ORIGINAL
ARTICLE
https://doi.org/10.35845/kmuj.2024.23635 ORIGINAL
ARTICLE
A nonsense variant of reactivation gene-1 leads to Omenn syndrome in a Pakistani family
Zara
Khalid ![]() 1,2,
Abeerah Zainab
1,2,
Abeerah Zainab![]() 1,3, Nida
Shafi
1,3, Nida
Shafi ![]() 1,4,
Summyah Niazi
1,4,
Summyah Niazi ![]() 1,5,
Sobia Humerah 1,6, Syed Irfan Raza
1,5,
Sobia Humerah 1,6, Syed Irfan Raza ![]() 1,7
1,7
|
1: Department of Biomedicine, National University of Science and Technology (NUST), Islamabad, Pakistan 2: Department of Biochemistry, Rawal Medical College, Islamabad, Pakistan 3: Department of Biochemistry, Islamic International Medical College, Rawalpindi, Pakistan 4: Department of Biochemistry, M. Islam Medical College, Gujranwala, Pakistan 5: Department of Physiology, Quetta Institute of Medical Sciences, Quetta, Pakistan 6: Department of Physiology, Al-Nafees Medical College, Isra University, Islamabad, Pakistan 7: Department of Biochemistry, HBS Medical College, Islamabad, Pakistan
Email
Date Submitted: March 15, 2024 Date Revised: August 28, 2024 Date Accepted: September 17, 2024 |
|
THIS ARTICLE MAY BE CITED AS: Khalid Z, Zainab A, Shafi N, Niazi S, Humerah S, Raza SI. A nonsense variant of reactivation gene-1 leads to omen Syndrome in a Pakistani family. Khyber Med Univ J 2024;16(3):201-6.https://doi.org/10.35845/kmuj.2024.23635 |
ABSTRACT
OBJECTIVE: To conduct clinical and genetic analysis in a patient with severe combined immunodeficiency disease (SCID).
METHODS: A 3.5-year-old female patient with chronic diarrhea, fever, and failure to thrive was examined at the Pakistan Institute of Medical Sciences, Islamabad. Ethical approval was obtained, and informed consent was secured for genetic studies. Flow cytometry was performed on blood samples to evaluate T, B, and NK cell concentrations. Genomic DNA was extracted from peripheral blood samples of the patient, her mother, and healthy siblings. Sanger sequencing of the RAG1 gene was conducted, followed by mutational analysis using BioEdit and bioinformatics tools for pathogenicity assessment.
RESULTS: The proband, the youngest girl born to first-cousin parents presented with intractable diarrhea with failure to thrive and skin and nappy rashes on clinical evaluation. The immunological assessment revealed total absence of B and T lymphocytes, normal NK cells (47% with 85% CD16 and CD56 respectively: No CD4, CD8 and CD19), and significant hypogammaglobulinemia (IgG;165 mg/dl, IgM; 9 mg/dl, IgA;8 mg/dl), while normal level of IgE (2 mg/dl). On the other hand, targeted Sanger sequencing of RAG1 exon 2 region revealed a new homozygous deleterious mutation (NM_000448.2: c.2876 G>A; p.Trp959*) in the RAG1 gene resulting in complete loss of function due to the production of truncated protein.
CONCLUSION: We identified a potentially pathogenic nonsense mutation in the RAG1 gene (c.2876 G>A; p.Trp959*) in a child with SCID. This finding emphasizes the significance of early detection for timely treatment and genetic counseling.
KEY WORDS: Immune phenotype (Non-MeSH); SCID (MeSH); Genetic Variation (MeSH); Sanger Sequencing (MeSH); Reactivation gene-1 (Non-MeSH).
INTRODUCTION
Severe combined immunodeficiency (SCID) comprises a genetically diverse group of disorders affecting the immune system and represents the highest mortality among inborn errors of immunity (IEI). 1 All forms of SCID share a common feature: defective T-cell production. While most also exhibit defective B-cell function, even when B cells are present, they cannot produce antibodies without T-cell assistance. Therefore, infants with SCID are highly susceptible to life-threatening infections.2 The incidence rate of severe combined immunodeficiency with total loss of B-cell and T-cell (B-/T-) type of SCID is one in 40,000 to 75,000 newborns.3 Clinical manifestations typically appear between 4 and 7 months of age and include recurrent infections, chronic diarrhea, and impaired growth.4 SCID patients are particularly vulnerable to community-acquired infections, often resulting in multi-organ dysfunction involving organs such as the lungs and liver.5
In humans, the adaptive immune system relies on V(D)J recombination to defend against various pathogens. This process is essential for generating unique antigen-binding sites on the surface of T cell receptors (TCRs) and B cell receptors (BCRs). During recombination, the variable (V), diversity (D), and joining (J) gene segments rearrange by breaking and rejoining specific DNA sequences that encode antigen receptors. This genetic reshuffling, known as segment recombination or isotype switching, is crucial for the diversity and adaptability of the immune response.6,7
Two proteins, RAG1 and RAG2 (Recombination Activating Gene 1 and 2), play a crucial role in the random recombination of the V, D, and J gene segments within the immunoglobulin and T-cell receptor loci. This process can potentially generate over 107 distinct antigen-binding sites.8,9 Mutations, both homozygous and heterozygous, in the RAG1 (OMIM 179615) and RAG2 (OMIM 179616) genes are associated with T-B-NK+ type autosomal recessive severe combined immunodeficiency (SCID) syndrome (OMIM 601457).9 The severity of the disease depends on the degree of impairment in recombinase activity.10 Patients with partial RAG function (<5%) may develop forms of leaky SCID (LS, OMIM 603554) or Omenn syndrome (OS, OMIM 603554). Individuals with leaky SCID or Omenn syndrome often present with symptoms such as atopic dermatitis, colitis, and mildly elevated IgE levels (hyper IgE syndrome).11-13 In this study, we enrolled a 3.5-month-old female patient admitted to the Pakistan Institute of Medical Sciences (PIMS), whose family originates from a remote village in Azad Jammu & Kashmir.
The patient presented with recurrent skin rashes, chronic diarrhea, and fever. The frequency of infections led us to hypothesize that she might have a compromised innate immune system, potentially due to a genetic variation. To investigate this, we conducted flow cytometry to evaluate her immune function and performed genetic testing to identify any mutations associated with the underlying disease etiology.
METHODS
A 3.5-year-old female patient was brought to the Pakistan Institute of Medical Sciences (PIMS), Islamabad, Pakistan with complaints of chronic diarrhea, fever, and failure to thrive. A thorough clinical examination was conducted by an expert panel of pediatricians to assess for possible bacterial, viral, or fungal infections, including pneumonia, chronic diarrhea, and respiratory tract infections. According to the patient's parents, she is the second child in their family to suffer from this condition.
The family was extensively interviewed, and a three-generation pedigree was constructed to determine the mode of inheritance.
Ethical statement: Prior to collecting detailed family and clinical histories, informed written consent was obtained from the parents for the genetic study and for the publication of the research findings in a peer-reviewed journal. The parents declined permission to publish facial features of the patient but allowed the sharing of images of the affected skin areas. Ethical approval for this study was obtained from the Institutional Review Board of HBS Medical College, Islamabad (IRB – EC20/4).
Operating Definitions
Consanguinity: Consanguinity (cousin marriage) was defined as father and mother of a patient being first or second cousin.14
Family pedigrees: A family chart related to genetics, that present relationships between family members and indicates which individuals have certain genetic pathogenic variants by using standardizing symbols. A family pedigree also indicates the pattern of inheritance of a disease allele.
Omen syndrome: Omen syndrome is a type of severe combined immune deficiency. By definition, patient should represent episodes of chronic diarrhea, failure to thrive, lymphadenopathy, hepatosplenomegaly etc.
Hypo-immunoglobinemia: Low level of circulatory immunoglobulin levels including IgG, IgA, IgM, IgE.
Laboratory Tests
Flow-cytometry:To evaluate the concentrations of circulating T cells, B cells, and natural killer cells (NKCs), a 2-3 ml whole blood sample was collected from the patient by expert phlebotomists using a "butterfly" needle, which includes flexible tabs and PVC tubing connected to the collection device. The sample was drawn into potassium ethylenediaminetetraacetic acid (K-EDTA) tubes (BD Vacutainer® EDTA Tubes, Becton Dickinson, UK). A complete blood count (CBC) was then performed using an automated Sysmex KX-21 Hematology Analyzer (Sysmex Corporation, Japan).
Genetic analysis: Peripheral blood samples (3-5 mL) were collected from the patient, her mother, and available healthy siblings. Genomic DNA was extracted from these samples using the GenElute™ Blood Genomic DNA Kit (Sigma-Aldrich, St. Louis, MO, USA). DNA purity and concentration were measured using a Nanodrop1000 spectrophotometer (Thermo Scientific, Wilmington, MA, USA). Given the patient’s symptoms, including chronic diarrhea, severe nappy rashes, and fever, we suspected an error in the V(D)J recombination process, which is typically mediated by reactivation gene 1 and 2 (RAG1 and RAG2), responsible for generating the TCR and BCR repertoire.
Following the exclusion mapping rule, we initially focused on sequencing RAG1 exons. Exon-specific primers were designed using PRIMER 3 software (http://bioinfo.ut.ee/primer3-0.4.0/), and polymerase chain reaction (PCR) was performed to amplify the selected exons from the patient’s DNA. The amplified DNA products were purified using a commercially available kit (Axygen, CA, USA), and Sanger sequencing was conducted by Alpha Genomics (Pvt) Ltd, Islamabad, following established protocols.15
Mutational analysis: RAG1 gene (OMIM 179615) sequencing data of patient and one parent was analyzed and compared with the corresponding control gene sequences available on Ensemble Genome Browser database (http://ensembl.org/index.html). In order to identify the nucleotide sequence variant BioEdit sequence alignment editor version 6.0.7 was used. To measure the pathogenicity of the identified variant, multiple bioinformatics tools were employed, including Polymorphism Phenotyping V2 (PolyPhen 2) and MutationTaster (http://www.mutationtaster.org/).
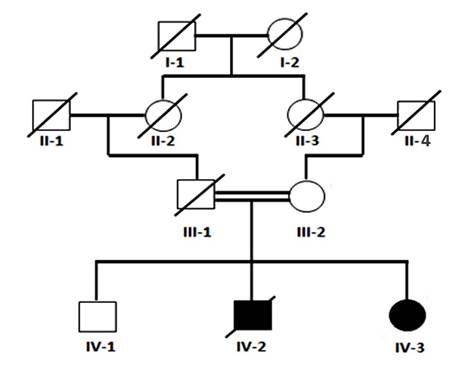
Figure 1: Pedigree of Family. Four generation Pakistani family pedigree segregating autosomal recessive type of severe combined immunodeficiency. Double lines indicate cousin marriages. Clear Square and circle symbols represent healthy subjects while filled symbols showing patients individuals, while a diagonal line indicates decease.
RESULTS
Clinical findings: In this study, we enrolled a 3.5-month-old female child, the second in birth order, diagnosed with SCID. According to the patient’s mother, the patient (III-2) is the second consecutive child in the family to suffer from body and nappy rashes, along with recurrent diarrhea. The first child, a male, died at six months of age due to recurrent pneumonia and skin rashes.
The female patient was first brought to the Department of Pediatrics at PIMS Hospital, Islamabad, at the age of three months, presenting with recurrent exudative skin eruptions, hyperthermia, and intractable diarrhea, leading to failure to thrive, along with persistent skin and nappy rashes. At 3.5 months, she was readmitted to the hospital with a poor general condition, presenting with persistent fever, pneumonia, diarrhea, and worsening cutaneous symptoms.
Given the family’s positive history of immune deficiency, the child had not received any vaccinations, including BCG. She was symptomatically treated by expert physicians with cyclosporine, corticosteroids, and supportive care.
Immunologic findings: Flow cytometry analysis revealed a total absence of B and T lymphocytes, while natural killer (NK) cells were within the normal range (47%). The distribution of T cells showed 13% CD3, 5% CD4, and 3% CD8. NK cell markers, including CD16 and CD56, were both normal at 85%, as shown in Table I. Evaluation of immunoglobulin levels and a complete blood count (CBC) confirmed hypogammaglobulinemia and lymphopenia. Detailed results are provided in Table I.
Serum immunoglobulin levels for both the patient and the mother are also shown in Table I. The patient demonstrated significantly lower levels of serum immunoglobulins compared to the mother.
Table I: Immunoglobulin levels & Lymphocyte subset in a case suspected of OMEN Syndrome
|
Variable |
Patients Results |
Mother Results |
Normal Range |
|
|
Immunoglobulin Levels |
IgG (mg/dl) |
165 mg/dl |
800mg/dl |
600–1,500 mg/dl |
|
IgM (mg/dl) |
9 mg/dl |
220mg/dl |
50–370 mg/dl |
|
|
IgA (mg/dl) |
8 mg/dl |
230mg/dl |
80–380 mg/dl |
|
|
IgE (mg/dl) |
2 mg/dl |
2.5mg/dl |
0–5 mg/dl |
|
|
Lymphocyte Subset Analysis |
WBC |
6500/µl |
10000/ µl |
4000-14000 cells/ µl |
|
Lymphocyte percentage |
1% |
40% |
44-76% |
|
|
Lymphocyte count |
65/µl |
3600/µl |
1760-10640cell/ µl |
|
|
CD16/56 |
0% (0) |
90% |
3-15% (160-950) cells/µl |
|
|
CD4:CD8 cells |
0 |
<1% 900mm3 |
1.48 –3.77 mm3 |
|
|
CD3+CD8 cells |
0% (0) |
34% (578) |
22-24% (500-1700) cells/µl |
|
|
CD19 cells |
0%(0) |
62% (1560) |
14-37% (610-2600) cells/µl |
|
|
CD3+ cells |
0% (0) |
75% (4425) cells/µl |
49-76% (1900-5900) cells/µl |
|
CIgG: Immunoglobulin G; IgM: Immunoglobulin M; IgA: Immunoglobulin A; IgE: Immunoglobulin E; CD: cluster of differentiation
Sequencing findings: Whole exome sequencing revealed mutation in few genes however patient’s detailed clinical features and flow-cytometry findings were clearly indicating the involvement of RAG1 or RAG2 deficiency. In order to find the pathogenic variant, targeted Sanger sequencing of the coding regions of RAG1 gene was performed. Sanger sequencing of RAG1 exon 2 region revealed a new homozygous deleterious variation (NM_000448.2: c.2876 G>A; p.Trp959*) in the RAG1 gene resulting in complete loss of function due to the production of truncated protein.
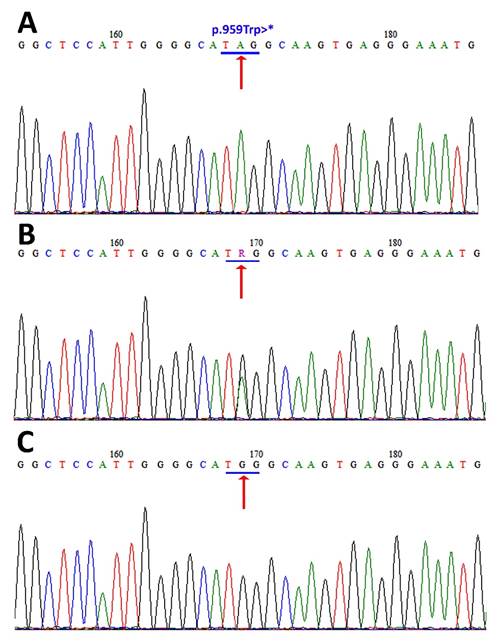
Figure 2: Sanger sequencing chromatogram of the family with RAG1 deficiency:. DNA sequence analysis of the RAG1 gene shows a substitution of G with A at nucleotide position 181 (c.2876 G>A; p.Trp959∗). (A) Arrow head showing homozygous mutant nucleotide in patient (IV-3). (B) Arrow head showing heterozygous nucleotide sequence in a carrier (III-2). (C) Arrow head showing position of homozygous wild-type nucleotide sequence in healthy family member (IV-1).
DISCUSSION
In this study, we identified a novel homozygous pathogenic mutation (c.2876 G>A; p.Trp959*) in the RAG1 gene through targeted Sanger sequencing, resulting in a truncated, non-functional protein. The patient exhibited severe immunodeficiency characterized by a total absence of B and T lymphocytes, consistent with RAG1 deficiency. These findings highlight the genetic basis of the patient's SCID phenotype.
In humans, the primary immunoglobulin (Ig) repertoire and the diverse range of antigen recognition receptors (TCRs) for B and T cells are generated through a molecular process known as V(D)J recombination.16 This process involves the assembly of V (variable), D (diversity), and J (joining) gene segments. V(D)J recombination is mediated by lymphoid-specific proteins encoded by recombination-activating genes (RAG1 and RAG2). RAG2 interprets the histone modifications on active DNA, while RAG1 identifies recombination signal sequences (RSS), and RAG2 directs RAG1 to introduce a nick in the DNA.17
Mutations in the RAG1 and RAG2 genes cause a spectrum of disorders, including severe combined immunodeficiency (T− B− NK+ SCID), Omenn syndrome (OMIM#603554), atypical or leaky SCID (AS), and delayed-onset combined immunodeficiency with granulomas and/or autoimmunity (CID-G/AI).
Omenn syndrome results from a partial loss of RAG gene function and presents with clinical features resembling SCID. It is an autosomal recessive form of SCID, clinically characterized by lymphadenopathy, frequent bacterial, viral, and fungal infections, severely disrupted lymph node architecture, lymphocyte depletion, hepatosplenomegaly, eosinophilia, and elevated IgE levels.11,12
In this study, the affected patient exhibited classic Omenn syndrome features, starting with a progressively diffuse erythrodermic scaly rash and recurrent infections, including diarrhea and pneumonia. By the age of three months, she developed generalized lymphadenopathy, hepatosplenomegaly, and continued to suffer from recurrent infections that were unresponsive to antibiotics but showed improvement with corticosteroid treatment. The use of corticosteroids, with or without cyclosporine, has been previously reported to significantly improve similar cases by suppressing autologous lymphocyte proliferation. All the clinical manifestations in this patient align with previously documented cases of Omenn syndrome caused by RAG mutations.1,11
DNA sequencing of the RAG-1 gene identified a previously reported loss-of-function missense mutation [c.2876 G>A; p.Trp959*].18 This mutation introduces a premature stop codon at position 959, resulting in a truncated RAG1 protein. According to the human genome mutation database, a total of 298 mutations—including missense, nonsense, small insertions/deletions, and large insertions/deletions—have been documented worldwide (Table II). The RAG1 protein consists of N-terminal, C-terminal, and catalytic core domains, with the identified nonsense mutation located in the C-terminal domain. This domain is essential for the interaction of the RAG1 protein with other proteins and plays a significant role in V(D)J recombination.19
CONCLUSION
SCID is a relatively rare genetic disorder characterized by a profound deficiency of both B and T lymphocytes, while natural killer (NK) cells remain present. Classical SCID and Omenn syndrome exhibit overlapping clinical features, including recurrent skin and respiratory infections, diarrhea, failure to thrive, and opportunistic infections. Diagnosis of SCID can be established in patients exhibiting these symptoms alongside positive NK cell counts. Confirmation of the diagnosis is achieved through genetic screening for RAG1 and RAG2 mutations.
ACKNOWLEDGEMENTS
We express our heartfelt gratitude to the participants, including patients, their parents, and healthy siblings, who took part in this study. Their invaluable contributions have been instrumental in enhancing our understanding of the underlying disease, Omenn syndrome.
REFERENCES
1. Shen J, Jiang L, Gao Y, Ou R, Yu S, Yang B et al. A novel RAG1 mutation in a compound heterozygous status in a child with Omenn syndrome. Front Genet 2019;10:913. https://doi.org/10.3389/fgene.2019.00913
2. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Cunningham-Rundles C, et al.. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol 2014;5:78108. https://doi.org/10.3389/fimmu.2014.00162
3.
Cossu F. Genetics of SCID. Ital J Pediatr 2010;36(1):76.
https://doi.org/10.1186/1824-7288-36-76
4. Ogando JCB, Gaytán AP, Becerra JCA, Cardona AÁ, Bezrodnik L, Borzutzky A, et al. Latin American consensus on the supportive management of patients with severe combined immunodeficiency. J Allergy Clin Immunol 2019;144(4):897-905. https://doi.org/10.1016/j.jaci.2019.08.002
5. Gennery A, Cant A. Diagnosis of severe combined immunodeficiency. J Clin Pathol 2001;54(3):191-5. https://doi.org/10.1136/jcp.54.3.191.
6. Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet 2011;45(1):167-202. https://doi.org/10.1146/annurev-genet-110410-132552.
7. Tonegawa S. Somatic generation of antibody diversity. Nature 1983;302(5909):575-81. https://doi.org/10.1038/302575a0.
8. Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem 2002;71(1):101-32. https://doi.org/10.1146/annurev.biochem.71.090501.150203
9. Thwaites DT. Novel insights into the biochemical mechanisms of RAG1 and RAG2 using mutational approaches (Doctoral dissertation, University of Leeds);2019.
10. Notarangelo LD, Kim M-S, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 2016;16(4):234-46. https://doi.org/10.1038/nri.2016.28
11. Rieux-Laucat F, Bahadoran P, Brousse N, Selz F, Fischer A, Le Deist F, et al. Highly restricted human T cell repertoire in peripheral blood and tissue-infiltrating lymphocytes in Omenn’s syndrome. J Clin Invest 1998;102(2):312-21. https://doi.org/10.1172/JCI332
12. Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell 1998;93(5):885-96. https://doi.org/10.1016/s0092-8674(00)81448-8
13. Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood 2001;97(1):81-8. https://doi.org/10.1182/blood.v97.1.81
14. Al-Mousa H, Al-Saud B. Primary immunodeficiency disease in highly consanguineous populations from Middle East and South Africa: Epidemiology, diagnosis and care. Front Immunol 2017;8:678. https://doi.org/10.3389/fimmu.2017.00678. eCollection2017
15. Raza SI, Nasser Dar R, Shah AA, Ahmad W. A novel homozygous nonsense mutation in the PVRL4 gene and expansion of clinical spectrum of EDSS1. Ann Hum Genet 2015;79(2):92-8.https://doi.org/10.1111/ahg.12094
16. Schatz DG. Transposition mediated by RAG1 and RAG2 and the evolution of the adaptive immune system. Immunol Res1999;19(2-3);169-82. https://doi.org/10.1007/BF02786485
17. McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, et al. Cleavage at a V (D) J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell 1995;3;83(3):387-95. https://doi.org/10.1016/0092-8674(95)90116-7
18. Lawless D, Lango Allen H, Thaventhiran J, Hodel F, Anwar R, Fellay J et al. Predicting the Occurrence of Variants in RAG1 and RAG2. J Clin Immunol 2019;39(7);688-01. https://doi.org/10.1007/s10875-019-00670-z
19. Santagata S, Gomez CA, Sobacchi C, Bozzi F, Abinun M, Pasic S, et al. N-terminal RAG1 frameshift mutations in Omenn's syndrome: internal methionine usage leads to partial V (D) J recombination activity and reveals a fundamental role in vivo for the N-terminal domains. Proc Natl Acad Sci 2000;97(26);14572-7. https://doi.org/10.1073/pnas.97.26.14572
AUTHORS' CONTRIBUTIONS Following authors have made substantial contributions to the manuscript as under:
ZK: Acquisition of data, drafting the manuscript, approval of the final version to be published AZ: Acquisition of data, critical review, approval of the final version to be published NS & SN: Study design, critical review, approval of the final version to be published SH: Concept and study design, acquisition, analysis and interpretation of data, drafting the manuscript, critical review, approval of the final version to be published SIR: Analysis of data, critical review, approval of the final version to be published
Authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. |
|
CONFLICT OF INTEREST Authors declared no conflict of interest, whether financial or otherwise, that could influence the integrity, objectivity, or validity of their research work.
GRANT SUPPORT AND FINANCIAL DISCLOSURE Authors declared no specific grant for this research from any funding agency in the public, commercial or non-profit sectors |
|
DATA SHARING STATEMENT The data that support the findings of this study are available from the corresponding author upon reasonable request |
|
|
|
KMUJ web address: www.kmuj.kmu.edu.pk Email address: kmuj@kmu.edu.pk |