![]() https://doi.org/10.35845/kmuj.2024.23631 CASE REPORT
https://doi.org/10.35845/kmuj.2024.23631 CASE REPORT
Intriguing challenging encounter of Clivus Chordoid Chordoma in a young girl: a case report
Fariha Sahrish ![]() 1, Alia Ahmad 1, Aysha Akram
1, Alia Ahmad 1, Aysha Akram ![]() 1, Amema Hafeez 1 Maira Jabbar
Chaudhary
1, Amema Hafeez 1 Maira Jabbar
Chaudhary ![]() 1
1
|
1. Department of Histopathology, Azra Naheed Medical College, Superior University, Lahore, Pakistan.
Email
Contact #: +92-323- 6578028
Date Submitted: March 05, 2023 Date Revised: September 08, 2024 Date Accepted: October 09, 2024 |
|
THIS ARTICLE MAY BE CITED AS: Sahrish F, Ahmad A, Akram A, Hafeez A, Chaudhary MJ. Intriguing challenging encounter of Clivus Chordoid Chordoma in a young girl: a case report. Khyber Med Univ J 2024;16(4):348-51. https://doi.org/10.35845/kmuj.2024.23631 |
ABSTRACT
Background: Clival Chordomas (CC) are rare, slow-growing tumors arising from primitive embryonic notochordal remnants, typically in the spheno-occiput, sacrococcygeal region, and clivus. They are slow-growing, locally aggressive tumors with limited metastatic potential and usually present with symptoms like headache, diplopia, and cranial nerve palsies. Diagnosis relies on magnetic resonance imaging (MRI) and histopathological examination with immunohistochemistry to distinguish them from chondrosarcoma.
Case presentation: A
five-year-old girl presented with a four-month history of diplopia and
right-sided hemiparesis. MRI revealed a hyperintense lobulated mass at the
skull base, compressing the spinal cord without cervical vertebral erosion.
Post-contrast imaging demonstrated heterogeneous enhancement. A comprehensive
baseline evaluation, including a metastatic assessment to exclude distant
spread, was conducted. Preoperative assessment was subsequently performed
during a tumor board meeting involving oncologists, pathologists,
anesthesiologists, and radiologists. Based on these evaluations, a gross total
resection of the tumor was planned. Differential diagnoses included chordoma
and chondrosarcoma, with ecchordosis physaliphora excluded radiologically.
Under general anesthesia, the tumor was successfully resected via a
transoronasal approach. Histopathological examination revealed lobules of large
cohesive cells within abundant myxoid stroma, consistent with a diagnosis of
chondroid chordoma. Immunohistochemical staining demonstrated diffuse
positivity for S100 and CK, effectively ruling out chondrosarcoma.
Conclusion: This case highlights the clinical,
radiological, and histopathological features of CC and emphasizes the
importance of immunohistochemical markers for accurate diagnosis. Complete
surgical resection remains the mainstay of treatment, complemented by
adjunctive radiotherapy for optimal management of this rare tumor.
Keywords: Chordoma (MeSH); Clivus (MeSH); Histological subtypes of Chordoma (Non-MeSH); Neoplasms (MeSH).
INTRODUCTION
Chordomas (CD) are uncommon malignant, slow-growing tumors of the axial skeleton, with a reported incidence of 0.08% cases per 100,000 persons per year in the United States of America (USA).1 These tumors show no sex predilection.2 Chordomas account for 1-4% of all bone tumors and 1% of intracranial tumors.3 CD arises in the spheno-occiput, sacrococcygeal region, and clivus due to the persistence of embryonic remnants of notochordal cells. Clival chordomas (CC) constitute one-third of these tumors.4
CC can be asymptomatic or present as headache, diplopia, dysphonia, hypoesthesia, ptosis, and cranial nerve palsies. In addition, patients can also develop complications i.e., cerebrospinal fluid rhinorrhea and brain abscess.5 The CT-Scan brain shows soft tissue masses and bone destruction. The high fluid content appears as hyperintense lesions on T1-weighted Magnetic Resonance Imaging (MRI). T2-weighted MRI shows a hypotense lesion with a moderate degree of contrast enhancement ruling out Ecchordosis Physaliphora radiologically.6 CC are locally aggressive but rarely metastasize. However, metastasis has been noted in other sites through hematogenous and lymphatic invasion, most commonly in the lungs, bones, and regional lymph nodes, with the lungs being the most frequently reported site.7
The World Health Organization (WHO) characterizes three types of CD: conventional/classical chordoma (CC), dedifferentiated chordoma (DC), and poorly differentiated chordoma (PDC). Chordoid chordoma (CCD) is a subtype of CC. The most important histological differential diagnosis of CCD is chondrosarcoma. Due to their similar clinical presentations, distinguishing between these entities poses challenges in clinical and perioperative diagnosis. CCD is of the hyaline and myxoid type, with atypical cytological and architectural features.
Immunohistochemical markers, including cytokeratin (CK), epithelial membrane antigen (EMA), brachyury, and S100, help differentiate CCD from chondrosarcoma. CK staining is seen in CC but absent in chondrosarcoma.1,3,5
Treatment protocols for CC typically include surgical resection, radiotherapy, and chemotherapy. However, CC shows resistance to neoadjuvant chemotherapy and radiotherapy. Radiotherapy is beneficial as an adjunct to surgical resection. Surgical approaches depend on the type and location of the tumor. Commonly used approaches include anterior endoscopic endonasal transsphenoidal resection (EETS), microscopic transoral (MTO) resection, and posterior approaches such as retrosigmoid (RS) and paramedian suboccipital (SO) resections.1,4,7-9
CASE REPORT
A five-year-old girl presented with diplopia and right-sided hemiparesis for four months in the neurosurgery department. There was no history of fever, weight loss, or a family history of cancer. The rest of her general physical examination and all baseline investigations were unremarkable. She was completely evaluated for any sensory and motor deficits in her upper limbs. Her MRI (T2-weighted) image showed a hyperintense lobulated mass present at the base of the skull with significant compression of the cord and no erosions of cervical vertebrae (Figure 1A). Her post-contrast (T1-weighted) image demonstrated heterogeneous enhancement at the base of the skull (Figure 1B).
Following a comprehensive panel of baseline investigations and a preoperative assessment during a tumor board meeting-comprising oncologists, pathologists, anesthesiologists, and radiologists-a gross total resection of the tumor was planned. The primary differential diagnoses were chordoma and chondrosarcoma, as ecchordosis physaliphora had been ruled out radiologically. Under general anesthesia, the tumor was completely resected via a transoronasal approach. The resected specimen, measuring 2 x 2 cm, consisted of grey-white, mucoid fragments, which were processed in their entirety for further evaluation.
Microscopic examination revealed lobules of large, cohesive physaliferous cells embedded within abundant myxoid stroma (Figure 1C). Additionally, areas containing benign-appearing chondrocytes within lacunae were observed. No cytological or architectural atypia was identified (Figure 1D). Immunohistochemical analysis demonstrated diffuse CK and S100 staining in tumor cells, effectively ruling out chondrosarcoma (Figure 1, E-F).
|
|
|
|
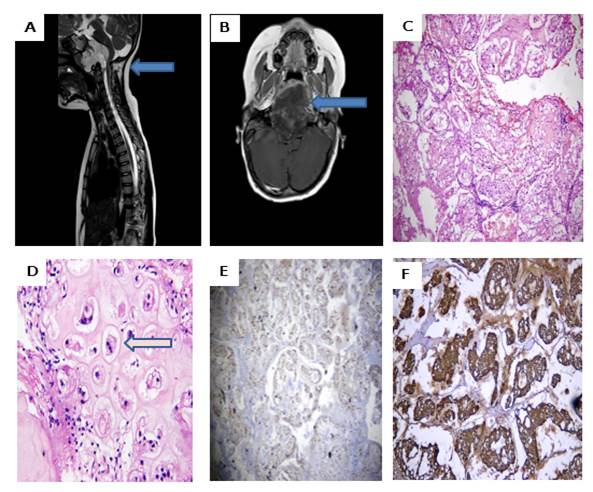
Figure 1: Radiological (MRI) and Histopathological features of Clival Chondroid Chordoma. A: A sagittal T2 weighted MRI image hyperintense lobulated mass present at the base of the skull with significant compressions of cord and no erosions of cervical vertebrae (Highlighted by arrows). B: Axial T1 weighted post-contrast slice demonstrates heterogeneous enhancement at the base of the skull (Highlighted by arrows). C: Photomicrograph of H&E stain shows lobules of large cohesive cells growing in an abundant myxoid stroma and physaliferous cells. D: Photomicrograph of H&E stain shows the benign-looking chondrocytes in myxoid stroma E: Cytoplasmic staining of CK in tumor cells. F: Strong and diffuse positivity of S100 in tumor cells.
DISCUSSION
A review of the literature indicates that CCD predominantly affects adult males. However, our index case of CCD in a five-year-old girl is consistent with findings by Rassi MS, et al., 5 who reported that 25.8% of CCD cases occur in patients under five years of age. The mean ages reported in various studies include 10.7 years by Rassi MS, et al.,5 10.6 years by Bilginer B, et al.,6 and a slightly higher mean age of 14.4 years by Zhai Y, et al.7 Ullah A, et al.,9 documented 5.8% of chordomas in patients younger than 19 years. In contrast, Gohar R, et al.,10 and Dickerson TE, et al.,11 identified CCD primarily in young adults aged 21-40 years. Among pediatric patients, females were more commonly affected, as noted by Rassi MS, et al.,5 Bilginer B, et al.,6 Zhai Y, et al.,7 and Erazo IS, et al.,8 Conversely, Ullah A, et al.,9 and Gohar R et al.10 reported a higher prevalence in males.
Regarding clinical features, our index case presented with diplopia, and right-sided hemiparesis. However, authors around the globe noted the varied clinical presentation i.e. headaches, diplopia strabismus, decreased visual defects/acuity, cranial nerve palsies commonly abducens nerve palsy, and quadriparesis.7,10 In contrast, Dickerson TE, et al., reported a metastatic CCD presented with abdominal pain and fullness.11
Regarding the histological subtype of CD, our case showed the morphology of CCD which is a subtype of CC, majority of authors reported the same subtype of CD. 5,8 As chondrosarcoma is a close differential diagnosis in the index study we applied CK and S100 Immunohistochemical staining. These stains are also reported by Rassi MS, et al., Zhai Y, et al., and Erazo IS, et al.5,7,8 However, Dickerson TE, et al., showed diffuse EMA and s100 positivity in the metastatic case of CCD. 11 It is crucial to note that Brachyury serves as a highly sensitive marker, and Ki67 a marker for the proliferation index is essential for prognosis.10 Regrettably, these pivotal markers are currently unavailable within our clinical setting.
Regarding metastasis, in the index study, there was no metastasis at the time of initial diagnosis, However, Bilginer B, et al., reported one case of tumor recurrence.6 Dickerson TE, et al., showed a diagnosed case of sacral CD which metastasized to the liver after 15 years of complete resection. 11 Following discharge, the patient was lost to follow-up, making it challenging to determine her survival. Nonetheless, Kamboh UA, et al., from Pakistan reported three cases of CCD in the spine, with one case demonstrating a consistently positive outcome. 12 There is limited available data on pediatric Clival CC from Pakistan, apart from our case report Kamboh UA, et al., and Imran M, et al., from Karachi reported CD in the spine.12,13
CONCLUSION
Clival chordomas are rare notochordal tumors primarily affecting the axial skeleton. This case highlights the clinical, radiological, and histopathological features of clival chordoma in a young girl successfully treated with gross total resection. The use of advanced immunohistochemical markers, including brachyury, was pivotal in ruling out differential diagnoses, ensuring accurate diagnosis. Complete surgical resection remains the cornerstone of treatment, with adjunctive radiotherapy playing a critical role in optimizing outcomes for this uncommon tumor.
REFERENCES
1. Pagella F, Ugolini S, Zoia C, Matti E, Carena P, Lizzio R, et al. Clivus pathologies from diagnosis to surgical multidisciplinary treatment. Review of the literature. Acta Otorhinolaryngol Ital 2021;41(Suppl 1):S42-S50. https://doi.org/10.14639/0392-100Xsuppl.1-41-2021-04
2. Ulici V, Hart J. Chordoma: a review and differential diagnosis. Arch Pathol Lab Med 2022;146(3):386-95. https://doi.org/10.5858/arpa.2020-58-RA
3. Rai R, Iwanaga J, Shokouhi G, Loukas M, Mortazavi MM, Oskouian RJ, et al. A comprehensive review of the clivus: anatomy, embryology, variants, pathology, and surgical approaches. Child's Nerv Syst 2018;34(8):1451-8. https://doi.org/10.1007/s00381-018-3875-x
4. Noya C, D’Alessandris QG, Doglietto F, Pallini R, Rigante M, Mattogno PP, et al. Treatment of clival chordomas: a 20-year experience and systematic literature review. Cancers (Basel) 2023;15(18):4493. https://doi.org/10.3390/cancers15184493
5. Rassi MS, Hulou MM, Almefty K, Bi WL, Pravdenkova S, Dunn I, et al. Pediatric clival chordoma: a curable disease that conforms to collins' law. Neurosurgery 2018;82(5):652-60. https://doi.org/10.1093/neuros/nyx254
6. Bilginer B, Türk CÇ, Narin F, Hanalioglu S, Oguz KK, Ozgen B, et al. Enigmatic entity in childhood: clival chordoma from a tertiary center’s perspective. Acta Neurochir 2015;157(9):1587-93. https://doi.org/10.1007/s00701-015-2510-9
7. Zhai Y, Bai J, Gao H, Wang S, Li M, Gui S, et al. Clinical features and prognostic factors of children and adolescents with clival chordomas. World Neurosurg 2017;98:323-8. https://doi.org/10.1016/j.wneu.2016.11.015
8. Erazo IS, Galvis CF, Aguirre LE, Iglesias R, Abarca LC. Clival chondroid chordoma: a case report and review of the literature. Cureus 2018;10(9):e3381. https://doi.org/10.7759/cureus.3381
9. Ullah A, Kenol GS, Lee KT, Yasinzai AQ, Waheed A, Asif B, et al. Chordoma: demographics and survival analysis with a focus on racial disparities and the role of surgery, a US population-based study. Clinic Transl Oncol 2024;26(1):109-18. https://doi.org/10.1007/s12094-023-03227-0
10. Gohar R, Rehman L, Javeed F, Bokhari I, Qasim A, Altaf S. Radiologic–histopathologic: correlation of intracranial tumors operated in a tertiary care hospital: a prospective study. Pak J Neurol Surg 2023;27(1):14-20. https://doi.org/10.36552/pjns.v27i1.813
11. Dickerson TE, Ullah A, Saineni S, Sultan S, Sama S, Ghleilib I, et al. Recurrent metastatic chordoma to the liver: a case report and review of the literature. Curr Oncol 2022;29(7):4625-31. https://doi.org/10.3390/curroncol29070367
12. Kamboh UA, Manzoor M, Gulshan A, Rauf A. The outcome of multidisciplinary treatment of spinal tumors: institutional experience of a tertiary care hospital. Pak J Neurol Surg 2022;26(4):589-96. https://doi.org/10.36552/pjns.v26i4.806
13. Imran M, Khan AA, Younus SM. Cervical chordoma involving C3/C4: A case report. J Pak Med Assoc 2016;66(12):1659-61.
Following authors have made substantial contributions to the manuscript as under: FS & AA: Identification and management of the case, drafting the manuscript, approval of the final version to be published AH, AK & MJC: Diagnosis of the case, management of the case, critical revision, approval of the final version to be published
Authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. |
|
CONFLICT OF INTEREST Authors declared no conflict of interest, whether financial or otherwise, that could influence the integrity, objectivity, or validity of their research work.
GRANT SUPPORT AND FINANCIAL DISCLOSURE Authors declared no specific grant for this research from any funding agency in the public, commercial or non-profit sectors |
|
DATA SHARING STATEMENT The data that support the findings of this study are available from the corresponding author upon reasonable request |
|
|
|
KMUJ web address: www.kmuj.kmu.edu.pk Email address: kmuj@kmu.edu.pk |