![]() https://doi.org/10.35845/kmuj.2024.23505
ORIGINAL ARTICLE
https://doi.org/10.35845/kmuj.2024.23505
ORIGINAL ARTICLE
Assessment of growth retardation among transfusion-dependent Thalassemia patients in Peshawar, Pakistan
Adil Zareef ![]() 1,
Farrukh Ansar 1,2, Aamir Ali Khan 3 , Muhammad Tariq
Masood
1,
Farrukh Ansar 1,2, Aamir Ali Khan 3 , Muhammad Tariq
Masood
Khan ![]() 4,
Farooq Shah 5
4,
Farooq Shah 5
|
THIS ARTICLE MAY BE CITED AS: Zareef A, Ansar F, Khan AA, Khan MTM, Ansari FM, Shah F. Assessment of growth retardation among transfusion-dependent Thalassemia patients in Peshawar, Pakistan. Khyber Med Univ J 2024;16(3):207-12. https://doi.org/10.35845/kmuj.2024.23505 |
ABSTRACT
OBJECTIVE: To investigate the relationship between repeated blood transfusions and growth retardation in children with transfusion-dependent Thalassemia in Peshawar, Pakistan.
METHODS: This cross-sectional study was conducted on transfusion-dependent thalassemia patients receiving treatment at the Fatimid Foundation, Peshawar Pakistan, between February and August 2022. Participants were categorized by age (<1 year, 1–2 years, and >2 years) to assess and measure growth patterns over six months.
RESULTS: Out of 93 children with thalassemia major, 55 (59.1%) were males and 38 (40.9%) were females. Mean age of the participants was 10.86 ± 5.72 years, and mean age at their first transfusion was 8.13 ± 5.78 months. Mean body mass index was 16.38 ± 1.82 kg/m². Short stature was observed in 49 patients (52.7%), of whom 57.1% (n=28) were male and 42.9% (n=21) were female. Serum ferritin levels were significantly elevated in patients with short stature, with 57.1% (n=28/49) having ferritin levels >4001 μg/L compared to 40.9% (n=18/44) with normal stature. Only 3.2% of patients had normal ferritin levels. The ROC analysis identified a ferritin cut-off of 1636 μg/L for predicting short stature (sensitivity 86%, specificity 68%). Growth assessment showed that 71% of the children were <50th percentile. Hemoglobin levels and early transfusion age were also associated with short stature, highlighting the impact of iron overload on growth.
CONCLUSION: This study highlights the detrimental effects of thalassemia on growth in transfusion-dependent children, primarily from iron overload and high ferritin levels, emphasizing the importance of better management strategies to prevent complications and promote healthy development.
KEYWORDS: Transfusion (MeSH); Thalassemia major (MeSH); Beta-Thalassemia (MeSH); Ferritins (Non-MeSH); Hyperferritinemia (MeSH); Iron Overload (MeSH); Growth Retardation (MeSH); Fetal Growth Restriction (MeSH); Growth Retardation, Intrauterine (MeSH); Body Mass Index (MeSH);
INTRODUCTION
Thalassemia major is the most prevalent monogenic hematologic disorder, affecting millions globally and resulting in thousands of deaths annually. Despite being one of the most common hereditary hemolytic disorders, thalassemia remains poorly understood regarding its genetic basis, categorization, biochemical abnormalities, and clinical manifestations. thalassemia arises from various interrelated genetic defects, leading to severe chronic conditions in different combinations.1 The disease spectrum ranges from being an asymptomatic carrier to severe hemolytic anemia, necessitating regular blood transfusions for survival. Managing transfusion-dependent thalassemia major (TDT) is challenging, as improper and repeated transfusions can lead to hemosiderosis, the primary cause of mortality. Nevertheless, iron overload complications can be mitigated through systematic protocols, regular follow-ups, and the use of effective iron chelators.2
Numerous studies conducted globally have demonstrated that recurrent transfusions in thalassemia patients result in elevated serum ferritin levels and excess iron accumulation. This iron overload can impair growth hormone function, contributing to developmental disorders such as growth retardation and short stature.3,4 For example, studies conducted in Malaysia and Yemen on prepubertal β-thalassemia major patients revealed a higher rate of growth failure compared to age- and gender-matched healthy children. In Yemen, growth retardation rates were significantly higher compared to other countries.5,6 A recent Brazilian study on children with transfusion-dependent β-thalassemia major found a higher prevalence of dental malocclusions, particularly Angle’s Class II type, indicating the broader impact of the disease on physical development.7 Additional research has revealed that thalassemia patients with elevated ferritin levels and iron overload from uncontrolled transfusions suffer from stunted growth and underdeveloped genitalia.8,9
A study at a transfusion center in Islamabad reported moderate to severe growth retardation in many frequently under-transfused β-thalassemia major patients.10 Similarly, research assessing the nutritional status of children with β-thalassemia major in Bahawalpur, Pakistan, found that most patients were underweight, with a significant association between nutritional status, age, and illness duration.11 Additionally, a significant reduction in Body Mass Index (BMI) was observed in Pakistani and Afghan β-thalassemia major patients compared to a control group, with both populations and genders showing notable declines in hematological parameters.12 Another cross-sectional study on β-thalassemia major patients in Karachi, Pakistan, revealed significant changes in biochemical profiles, including bone abnormalities.13 In Pakistan, the β-thalassemia carrier rate is estimated at 5.0–8.0%, making it a common hereditary blood disorder. Homozygous affected children rely on regular and expensive blood transfusions for survival. This highlights the importance of preventive strategies like mutation detection, genetic counseling, prenatal diagnosis, carrier screening, and increasing public awareness.14
With limited resources and the lack of universal healthcare services, thalassemia is poised to present a growing challenge in Pakistan.8 This study was planned to assess the association between transfusion-dependent thalassemia and growth impairment in children, along with related complications. Specifically, we focused on the elevated blood ferritin levels (>500 ng/mL) commonly seen in poorly managed transfusion-dependent thalassemia patients, which contribute to growth retardation. Key contributing factors include inadequate pre-transfusion hemoglobin levels, delayed or insufficient chelation therapy, and poor treatment compliance. By addressing these issues, our study seeks to fill a critical gap in understanding the impact of high ferritin levels on growth in children with thalassemia in Pakistan. The findings will provide valuable insights for optimizing treatment protocols, improving patient outcomes, and reducing the long-term complications associated with thalassemia.
METHODS
This cross-sectional study was designed to provide a standardized quantitative assessment and measurement of growth patterns in transfusion-dependent thalassemia patients. The research was conducted at the Fatimid Blood Bank & Foundation in Hayatabad, Peshawar, over a six-month period from February to August 2022. Using a purposive sampling technique, we recruited 93 confirmed transfusion-dependent thalassemia major patients receiving treatment at the Fatimid Foundation, Peshawar-Pakistan. Patients who declined participation were excluded from the study.
Ethical approval was obtained from the Fatamid Foundation (228/Research & IN. S/01/FFP), and a comprehensive data collection questionnaire was shared with the participants. Informed consent, emphasizing the confidentiality of collected data, was obtained from both patients and guardians of minors. To maintain confidentiality, each patient was assigned a different serial number within their respective categories. Calibrated scales were utilized for standardized accuracy in measurements.
Ferritin level analysis was carried out using COBASE e 411 Roche (Hitachi), and CBC analysis was performed using the SYSMAX HB automatic device.
Data were systematically recorded on standardized Performa sheets and analyzed using IBM SPSS version 22. Descriptive statistics, including means and standard deviations, were calculated for continuous variables such as age, while frequencies, percentages, and percentiles were computed for categorical data. Significance between means was assessed. Additionally, Pearson’s correlation was employed for analyzing the correlation between continuous predictor values, and Chi-Square tests were used for categorical variables.
RESULTS
Among the 93 transfusion-dependent thalassemia major patients, 55 (59.1%) were male, and 38 (40.9%) were female. Short stature was observed in 49 patients (52.7%), while 44 (47.3%) had normal stature (Table I). Specifically, 50.9% of the males (n=28/55) had short stature, compared to 55.3% of the females (n=21/38). Of the patients with short stature, 57.1% (n=28/49) were male, and 42.9% (n=21/49) were female.
Table I: Percentage of baseline characteristics of the study participants
|
Variable |
Normal stature (n=44) |
Short Stature (n=49) |
Total (n=93) |
|
|
Frequency (%) |
Frequency (%) |
Frequency (%) |
||
|
Gender |
Male |
27 (61.36%) |
28 (57.14%) |
55 (59.14%) |
|
Female |
17 (38.64%) |
21 (42.85%) |
38 (40.86%) |
|
|
Ferritin (μg/L) |
Normal Range |
2 (4.54%) |
1 (2.04 %) |
3 (3.22%) |
|
337 to 1000 |
6 (13.64%) |
2 (4.08%) |
8 (8.60%) |
|
|
1001 to 2500 |
9 (20.46%) |
9 (18.36%) |
18 (19.36%) |
|
|
2501 to 4000 |
9 (20.46%) |
9 (18.36%) |
18 (19.36%) |
|
|
> 4001 |
18 (40.90%) |
28 (57.14%) |
46 (49.46%) |
|
|
Hemoglobin (gm/dl) level before transfusion |
<5-8 |
15 (34.09%) |
17 (34.69%) |
32 (38.70%) |
|
>8-10 |
19 (43.18%) |
26 (53.06%) |
45 (48.83%) |
|
|
>10 |
10 (22.72%) |
6 (12.24%) |
16 (17.20%) |
|
|
Age (years) at first transfusion |
<1 |
32 (72.27%) |
43 (87.75%) |
75 (80.46%) |
|
1-2 |
9 (20.45%) |
6 (12.24%) |
15 (16.12%) |
|
|
> 2 |
3 (9.67%) |
0 (0%) |
3 (3.22%) |
|
Serum ferritin levels were generally higher in patients with short stature than in those with normal stature. Twenty-eight (57.1%) patients with short stature had ferritin levels exceeding 4001 μg/L, compared to 18 (40.9%) patients with normal stature. Only 3 (3.2%) of all transfusion-dependent thalassemia major patients had serum ferritin levels within the normal range. A Receiver Operating Characteristic (ROC) curve analysis determined a ferritin cut-off of 1636 μg/L for predicting short stature, with a sensitivity of 86% and specificity of 68% (Figure 1, Table II).
Table II: Youden index J, sensitivity, and specificity
|
Variable |
Value |
|
Youden Index J |
0.540 |
|
Area under the curve |
.636 |
|
p-value |
.024 |
|
Cut-off value of Ferritin |
1636 |
|
Sensitivity |
86% |
|
Specificity |
68% |
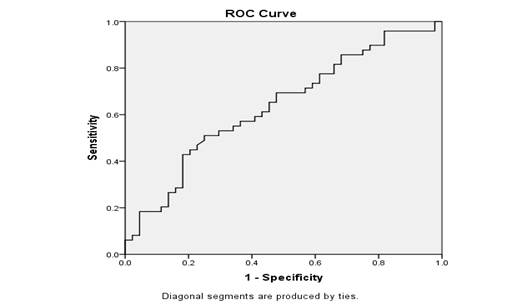
Figure 1: Receiver operating characteristic curve of serum ferritin levels with short stature
Hemoglobin levels before transfusion were also assessed. Nearly half of the patients (n=45; 48.83%) had pre-transfusion hemoglobin levels between 8-10 g/dL, while 38.70% had levels between 5-8 g/dL. Patients with short stature were more likely to have lower pre-transfusion hemoglobin levels compared to those with normal stature (Table I).
The majority of patients (n=75; 80.46%) received their first transfusion before the age of one year. A greater proportion of patients with short stature (n=43; 87.75%) had earlier transfusions compared to those with normal stature (n=32; 72.27%). Only 3 patients (3.22%) received their first transfusion after the age of two years (Table I).
Correlation analysis revealed a significant association between higher serum ferritin levels and short stature (r = -0.247, p = 0.017) and an inverse correlation with growth chart percentiles (r = -0.258, p = 0.012). Additionally, there was a positive correlation between ferritin levels and patient age (r = 0.212, p = 0.041).
Logistic regression analysis identified younger age and age at first transfusion as significant predictors of short stature in transfusion-dependent thalassemia patients. The Wald value for age was 8.28 (p = 0.004), while for age at first transfusion, it was 3.51 (p = 0.061) (Table III).
Table III: Logistic regression analysis
|
Variables |
B |
SE |
Wald |
p-value |
Exp (B) |
|
Age of patient |
.233 |
.081 |
8.28 |
.004 |
.792 |
|
Age at first transfusion |
.114 |
.061 |
3.51 |
.061 |
1.12 |
|
Growth percentile |
.010 |
.016 |
.394 |
.530 |
1.01 |
In our study, growth percentile data revealed that 66 children (71%) were below the 50th percentile on the growth chart, 23 (24.7%) were between the 50th and 90th percentiles, and only 4 (4.3%) were above the 90th percentile. In terms of splenectomy status, 8 patients (8.6%) had undergone splenectomy, while 85 (91.4%) had not.
Pearson’s correlation analysis was done to find out any potential correlation between serum ferritin levels and other levels. Analysis showed that an increase in serum ferritin levels were correlated with short stature (r = -.247, p-value = .017). A positive correlation was found between serum ferritin levels and age (r = .212, p-value = .041) whereas negative correlation was reported between serum ferritin levels and the age of first transfusion (r = -.056, p-value = 596) and growth chart percentiles (r = -.258, p-value = .012).
DISCUSSION
In this cross-sectional study conducted in Peshawar, Pakistan, we explored the link between transfusion-dependent thalassemia and growth retardation in children. The study included 93 thalassemia major children, both male and female, with a mean age of 10.86 ± 5.72 years. The severity of the condition was reflected by the early initiation of transfusions, with the mean age of the first transfusion being 8.13 ± 5.78 months. Our results indicated that while early transfusions are lifesaving, they contribute to iron overload and elevated ferritin levels, leading to multiple endocrinopathies and growth impairments. The study revealed a significantly low mean body mass index (BMI) of 16.24 ± 1.96 for males and 16.58 ± 1.59 for females. Furthermore, 96.8% of children exhibited abnormally high serum ferritin levels.
Thalassemia major patients are expected to face fatal outcomes within months of diagnosis without timely transfusions or transplantation. In many developing countries, long-term transfusion and chelation therapy present significant challenges, imposing an unsustainable healthcare burden.12 In Pakistan, the most recent data from 2018 estimates 9.8 million thalassemia carriers, with a carrier rate of 5-7%. Given the country's low human development index ranking of 146, thalassemia poses considerable strain on already limited healthcare resources.8 Iron overload-induced endocrinopathies, commonly occurring within the first decade of life, result in growth disturbances and thyroid dysfunction.15
In our study, 8 patients had undergone splenectomy, a common intervention for splenomegaly caused by extramedullary hematopoiesis and increased red blood cell destruction, which heightens transfusion needs. We also observed a significantly low mean BMI of 16.24 ± 1.96 for males and 16.58 ± 1.59 for females, pointing to the prevalence of stunted growth and growth retardation. These findings align with previous research conducted on the Iranian population in 2004.16 Similarly, another study on Pakistani and Afghan β-thalassemia major patients reported a substantial decline in BMI compared to control groups.10
Our study revealed that 91 out of 93 (%age) children with thalassemia major exhibited abnormally high serum ferritin levels, a consequence of long-term reliance on blood transfusions. Similar to findings by Shalitin et al. in 2005, elevated serum ferritin levels were associated with impaired growth and delayed puberty, particularly in thalassemia major patients.17 High ferritin levels throughout the first decade of life strongly indicated short stature in these children. The frequent need for transfusions often results in iron overload, leading to complications such as stunted growth, facial deformities, and a protruding abdomen. In this study, we measured key anthropometric parameters including height, weight, head circumference, abdominal girth, and upper arm circumference to calculate growth chart percentiles. Among the 93 children, 66 (71%) were below the 50th percentile, 23 (24.7%) were below the 90th percentile, and only 4 (4.3%) were above the 90th percentile. The primary aim was to assess growth retardation in transfusion-dependent thalassemia children. We observed that continuous transfusions lead to iron accumulation and elevated serum ferritin levels, resulting in developmental delays, delayed puberty, physical deformities, and various endocrinopathies, including hypogonadism, hypothyroidism, diabetes mellitus, liver disease, and cardiovascular disease-all of which warrant further investigation.17
To alleviate these challenges, it is recommended to explore alternative strategies that reduce the dependence on transfusions, such as the use of Hydroxyurea, which stimulates fetal hemoglobin production and has been proven to decrease transfusion frequency in children with β-thalassemia.18 Additionally, ensuring the affordability and consistent availability of chelating agents in the market is crucial to maintaining an uninterrupted supply chain. Strict adherence to established management protocols should also be enforced to optimize patient outcomes. Moreover, the active involvement of non-governmental organizations and donors is essential to securing funding for high-cost treatments at the local level, helping to alleviate the financial burden on affected families.
Limitation of the study
The limited sample size and cross-sectional design of our study restrict the generalizability of the findings. Furthermore, the study did not assess hypogonadism, a significant factor contributing to growth failure in beta thalassemia major. A comprehensive evaluation of other factors, such as hormonal status, endocrinopathies, and their relationship with serum ferritin levels, is essential for a better understanding of growth impairment in these patients.
Future directions: Research should focus on evaluating the long-term efficacy of Hydroxyurea (HU) as a potential therapy to reduce the need for transfusions in thalassemia patients. In addition, further studies are needed to explore other therapeutic options and interventions that can alleviate the negative impact of iron overload and enhance the quality of life in thalassemia patients. Advancements in genetic screening, early diagnosis, and personalized treatments will be crucial in improving the long-term management of this chronic condition.
CONCLUSION
Our study highlights the significant growth retardation observed in transfusion-dependent thalassemia patients, primarily driven by iron overload and elevated serum ferritin levels. These findings emphasize the importance of enhancing management strategies to prevent iron overload, promote normal growth, and minimize long-term complications in transfusion-dependent thalassemia patients.
ACKNOWLEDGMENT
We would like to acknowledge Fakeha Meraj Ansari for her valuable contributions to the data collection process in this research project.
REFERENCES
1. Valentine WN, Neel JV. Hematologic and genetic study of the transmission of thalassemia:(cooley's anemia; mediterranean anemia). Arch Intern Med (Chic) 1944;74(3):185-96. https://doi.org/10.1001/ARCHINTE.1944.00210210032005.
2. Bayanzay K, Alzoebie L. Reducing the iron burden and improving survival in transfusion-dependent thalassemia patients: current perspectives. J Blood Med 2016;7:159-69. https://doi.org/10.2147/JBM.S61540.
3. Rathaur KV, Imran A, Pathania M. Growth pattern in thalassemic children and their correlation with serum ferritin. J Family Med Prim Care 2020;9(2):1166-9. https://doi.org/10.4103/jfmpc.jfmpc95119.
4. Atmakusuma TD, Hasibuan FD, Purnamasari D. The correlation between iron overload and endocrine function in adult transfusion-dependent beta-thalassemia patients with growth retardation. J Blood Med 2021;12:749-53. https://doi.org/10.2147/JBM.S325096.
5. Hamidah A, Arini MI, Zarina AL, Zulkifli SZ, Jamal R. Growth velocity in transfusion dependent prepubertal thalassemia patients: results from a thalassemia center in Malaysia. Southeast Asian J Trop Med Public Health 2008;39(5):900-5.
6. Al-Tayar AAK, Al-Zaazaai AAM. Growth pattern among Yemeni children suffering from Β thalassemia major in relation to serum ferritin the Yemeni Society for thalassemia and genetic blood disorder - Sana’a Yemen. J Endocrinol Diab 2019;6(3):1-7. https://doi.org/10.15226/2374-6890/6/3/001136.
7. Wonke, B, De Sanctis V. Clinical aspects of transfusional iron overload. Rev Clin Exp Hematol 2000;4(4):322-36. https://doi.org/10.1046/j.1468-0734.2000.00023.x.
8. Moiz B, Habib A, Sawani S, Raheem A, Bilal H, Gangwani M. Anthropometric measurements in children having transfusion-dependent beta thalassemia. Hematology 2018:23(4):248-52. https://doi.org/10.1080/10245332.2017.1396044.
9. Shahid Z, Hassan S, Ghazanfar S, Kaneez M, Khan MS, Tariq HT, et al. Investigating the role of ferritin in determining sexual underdevelopment in beta-thalassemia major patients: a cross-sectional analysis from Pakistan. Cureus 2021:13(6):e15572. https://doi.org/10.7759/cureus.1557.2
10. Sheikh MA, Shakir MU, Shah M. The assessment of nutritional status of children with beta thalassemia major with body mass index. Pak J Med Health Sci 2017:11(1):262-5.
11. Sultan S, Irfan SM, Ahmed SI. Biochemical markers of bone turnover in patients with β‐thalassemia major: a single center study from Southern Pakistan. Adv Hematol 2016;2016:5437609. https://doi.org/10.1155/2016/5437609
12. Muhammad R, Shakeel M, Rehman S, Lodhi MA. Population-based genetic study of β-thalassemia mutations in Mardan Division, Khyber Pakhtunkhwa Province, Pakistan. Hemoglobin 2017;41(2):104-9. https://doi.org/10.1080/03630269.2017.1330210
13. Jaffri SA, Irfan SM, Sultan S, Usman SM, Nadeem S. Bone Mineral Density among adolescent's patients with β-thalassemia major. Int J Endorsing Health Sci Res; 2021; 9 (1):42-7. https://doi.org/10.29052/IJEHSR.v9.i1.2021.42-4
14. Beatrix W, Sanctis VD. Clinical aspects of transfusional iron overload. Rev Clin Exp Hematol 2000; 4(4):322-36. https://doi.org/10.1046/j.1468-0734.2000.00023.x
15.Yousefian S, Mirialiabad G, Saleh R, Khedmati M. Association of Body mass index and serum ferritin level in pediatrics with Beta-thalassemia major disease. Iranian J Pediatric Hematol Oncol 2022;12(1):34-40. https://doi.org/10.18502/ijpho.v12i1.8359
16. Shalitin, S, Carmi D, Weintrob M, Philip M, Miskin H, Kornreich L, et al. Serum ferritin level as a predictor of impaired growth and puberty in thalassemia major patients. Eur J Hematol 2005;74(2):93-100.https://doi.org/10.1111/j.16000609.2004.00371. x
17. Hatamleh MI, Chenna VSH, Contractor H, Krishna Mohan GV, Tirumandyam G, Dammas N, Khan MW, Hirani S. Efficacy of Hydroxyurea in Transfusion-Dependent Major β-Thalassemia Patients: A Meta-Analysis. Cureus. 2023 ; 15(4):e38135. https://doi.org/10.7759/cureus.38135
Following authors have made substantial contributions to the manuscript as under:
AZ: Conception
and study design, drafting the manuscript, critical review, approval of the
final version to be published
Authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. |
|
CONFLICT OF INTEREST Authors declared no conflict of interest, whether financial or otherwise, that could influence the integrity, objectivity, or validity of their research work.
GRANT SUPPORT AND FINANCIAL DISCLOSURE This study was partially supported by Khyber Medical University Publication Fund (Reference No. DIR/ORIC/Ref/24/00085) |
|
DATA SHARING STATEMENT The data that support the findings of this study are available from the corresponding author upon reasonable request |
|
|
|
KMUJ web address: www.kmuj.kmu.edu.pk Email address: kmuj@kmu.edu.pk |